


Bs CK2 Nguyễn Ngọc Văn Khoa - Khoa Cấp Cứu
Kháng insulin (KI) là một biến chứng thường thấy của bệnh thận mạn (BTM) giai đoạn cuối dựa trên thử nghiệm kẹp đẳng đường-cường insulin. Kháng insulin cũng có mặt trong bệnh thận mạn giai đoạn đầu, ngay cả khi tốc độ lọc cầu thận nằm trong phạm vi bình thường. Tốc độ thanh thải glucose tương quan nghịch với nồng độ creatinine huyết thanh và tương quan thuận với độ thanh thải creatinin. Nhiều nghiên cứu trên diện rộng sử dụng dữ liệu từ các nguy cơ xơ vữa động mạch trong cộng đồng đã xác nhận hội chứng chuyển hóa dự đoán được nguy cơ mắc bệnh thận mạn và nhận thấy kháng insulin có liên hệ với bệnh thận mạn và sự suy giảm nhanh chóng chức năng thận. Vậy cơ chế KI ở bệnh thận mạn như thế nào?
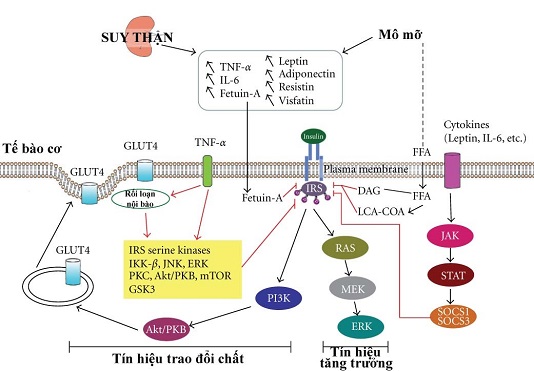
Kháng insulin ở bệnh thận mạn tính
Chú thích: Suy thận dẫn đến tình trạng viêm mô mỡ toàn thân và tăng cấp độ chuyển hóa mỡ trong huyết tương. TNF-α kích hoạt mô mỡ phân hủy, tạo ra các axit béo tự do. Ở các tế bào cơ, FFAs kích hoạt các yếu tố phiên mã, chẳng hạn như thụ thể kích hoạt bởi peroxisome proliferator (peroxisome proliferator-activated receptor - PPAR), và tạo ra các sứ giả bao gồm diacylglycerol (DAG) và chuỗi acyl-CoA (LCA-CoA), sẽ dẫn đến kích hoạt protein kinase C (PKC) và hủy photphoryl hóa của thụ thể insulin nền (IRS) -1/2. Ở các tế bào cơ, TNF-α kích hoạt một loạt các kinase bao gồm IKK-β, c-Jun NH2-terminal kinase (JNK), kinase ngoại bào quy định tín hiệu (ERK), protein kinase C (PKC), Akt (PKB), rapamycin ở người (mTOR), và glycogen synthase kinase 3 (GSK3) chịu trách nhiệm cho sự phosphoryl hóa của thụ thể insulin (InsR) và IRS-1 trên dư lượngserine / threonine. Sự ức chế chức năng của IRS-1 sẽ chặn Akt dẫn đến hấp thụ cytosol glucose transporter 4 (GLUT4). Fetuin-A cũng có thể ức chế IRS và gây viêm cấp thấp. IL-6 chịu trách nhiệm cho sự cảm ứng các bộ triệt khác nhau của protein tín hiệu cytokine (SOCS) thông qua các lộ trình tín hiệu Janus kinase / tín hiệu đầu dò và hoạt hóa phiên mã (Jak / STAT). SOCS sẽ ức chế IRS-1/2 và protein kinase A. Rối loạn lưới nội chất có vẻ là một yếu tố nữa liên kết viêm với kháng insulin ở cấpđộ phân tử. Dòng đỏ minh họa cơ chế của các yếu tố liên quan đến suy thận trong KI.
1. Kháng insulin thúc đẩy tổn thương thận
KI thúc đẩy bệnh thận bằng cách làm mất cân bằng huyết động tại thận thông qua các cơ chế như kích hoạt của hệ thống thần kinh giao cảm, ứ đọng muối, giảm Na +, K +-ATPase hoạt lực, và tăng mức lọc cầu thận. Rối loạn lưới nội bào dường như là yếu tố liên kết viêm và KI ở cấp độ phân tử. Việc ức chế các tín hiệu insulin thông qua phosphoryl hóa cơ chất thụ thể insulin do chất hoạt hóa c-Jun kinase N-terminal (JNK) đóng một vai trò quan trọng. Rối loạn lưới nội chất thận có liên quan đến tổn thương tế bào có chân gây ra bởi đạm niệu và biến đổi nephrin N-glycosyl hóa ở tế bào có chân, là yếu tố cơ bản trong sinh bệnh học của đạm niệu, và tham gia vào sinh lý bệnh của tổn thương thận mạn tính với tổn thương ống - kẻ thận. KI và các cytokine viêm có thể phần nào chịu trách nhiệm cho việc mở rộng tế bào cạnh cầu thận, làm dày màng nền tế bào, tổn thương tế bào có chân, và mất khe lỗ màng ngăn hoàn toàn cuối cùng dẫn đến chứng xơ cứng tiểu cầu thận và tổn thương ống - kẻ thận. Megalin, một thụ thể nội bào, làm trung gian cho việc tái hấp thụ các chất dinh dưỡng và vận chuyển protein gắn kết vitamin trong các bộ lọc cầu thận thông qua sự tương tác với các phân tử khác nhau trong các tế bào biểu mô ống lượn gần. Trong hội chứng chuyển hóa hoặc rối loạn lipid máu, các axit béo tự do được gửi đến tế bào biểu mô ống lượn gần với protein vận chuyển như albumin hoặc protein gắn kết axit béo ở gan. Trao đổi chất quá tải ở ống lượn gần được kích hoạt để xuất hiện các cytokin tiền viêm, chẳng hạn như MCP1 và TNFα, và dẫn đến chuyển tiếp biểu mô - trung mô.
KI có thể dẫn đến tăng LDL cholesterol và góp phần tăng triglyceride máu. Apolipoprotein B giàu triglyceride rõ ràng thúc đẩy sự tiến triển của suy thận. Mức triglyceride cao là một yếu tố nguy cơ cho đạm niệu phát triển.
2. Các yếu tố ảnh hưởng đến kháng insulin trong bệnh thận mạn
Thận đóng một vai trò quan trọng trong chuyển hóa insulin. Ước tính có khoảng 30- 80% insulin trong hệ tuần hoàn được lọc qua thận. Insulin có trọng lượng phân tử 6000 Dalton và được tự do lọc qua thận. Khi suy thận, giảm mức lọc cầu thận, giảm chuyển hóa thận của insulin dẫn đến kéo dài thời gian và nồng độ đỉnh của nó trong huyết tương, do đó bệnh nhân muốn uống đường. Mặc dù vậy biểu đồ đường huyết là bất thường, chứng minh cho sự KI ở tế bào. Một số yếu tố trong BTM góp phần làm KI như nồng độ urê huyết cao, toan chuyển hóa, thiếu vitamin D.
KI trong suy thận mạn tồn tại chủ yếu ở ngoại vi. Bởi vì mô mỡ thanh lọc dưới 2% tải lượng đường huyết, mô cơ là nơi biểu hiện chính về KI ở bệnh nhân bệnh thận giai đoạn cuối. Sản xuất glucose ở gan không tăng và được giữ ở mức bình thường để đáp ứng insulin ở bệnh nhân bệnh thận giai đoạn cuối. KI có thể không dẫn đến một sự gia tăng bù tiết insulin, so với bệnh nhân không mắc bệnh thận. Ngược lại với mô hình động vật với lượng chất béo hấp thụ gây ra bởi KI cao, KI trước tiên biểu hiện ở gan và sau đó trong mô mỡ trắng, trong khi cơ xương vẫn còn nhạy cảm với insulin. Một khiếm khuyết sau thụ thể được công nhận là khiếm khuyết chính trong bệnh thận mạn. Một nghiên cứu đã chứng minh rằng hoạt hóa của thụ thể insulin IRS-1 liên kết phosphoinositol (PI) 3-kinase (K) đã bị ức chế. Các cơ chế tiềm năng cho việc giảm hoạt hóa thụ thể IRS-1 liên kết (PI) 3-kinase (K) là cảm ứng của các tiểu đơn vị điều tiết P85 PI3-K, nhưng một cơ chế khác cũng có thể liên quan. Chức năng chính của IRS-PI3 K-Akt gắn liền với viêm mạn tính, nhiễm toan chuyển hóa, vitamin D và tình trạng hormone tuyến cận giáp, thiếu máu, chất độc urê máu, và chuyển hóa mỡ.
2.1. KI kết hợp với viêm nhiễm trong bệnh thận mạn
Một quá trình phức tạp những biến đổi về dinh dưỡng và trao đổi chất làm nền tảng cho suy thận mạn, bao gồm cả viêm mạn, rối loạn oxy hóa, KI, và suy dinh dưỡng protein năng lượng. Giống như các bệnh mạn tính khác, BTM có biểu hiện viêm cấp độ nhẹ đánh dấu bằng nồng độ các cytokine tiền viêm như protein phản ứng C (CRP), yếu tố hoại tử khối u (TNF-α), inerleukin-6 (IL-6), và interleukin-1 beta (IL-1β). Viêm và rối loạn oxy hóa là điều hiển nhiên trong giai đoạn đầu của suy thận và cũng gây ra KI, chủ yếu thông qua tăng sản xuất các cytokine tiền viêm.
Trong sự hiện diện của insulin, các thụ thể insulin phosphoryl IRS, được liên kết với sự kích hoạt của hai lộ trình truyền tín hiệu chính: phosphatidylinositol 3-kinase (PI3 K) -AKT / protein kinase B (PKB), lộ trình chịu trách nhiệm đối với hầu hết các các hoạt động trao đổi chất như vận chuyển glucose, và enzym kích hoạt phân bào Ras (Ras-mitogen-activated protein kinase -MAPK), lộ trình quy định biểu hiện gen và kết hợp với lộ trình PI3 K để kiểm soát sự tăng trưởng tế bào và sự phân hóa tế bào. Trong các mô hình động vật của bệnh thận mạn, hoạt động PI3 K giảm còn sự chết tế bào (apoptosis) và lộ trình ATP-ubiquitin-proteasome được kích hoạt.
TNF-α gây ra kháng insulin thông qua các cơ chế trực tiếp hoặc gián tiếp (hình 1.1). TNF-α có thể gây ra phosphoryl hóa IRS-1 thông qua hoạt hóa kinase serine bao gồm IKK-β, c-Jun kinase NH2 ở đầu tận cùng (c-Jun NH2-terminal kinase -JNK), tín hiệu ngoại bào điều tiết kinase (extracellular signal-regulated kinase -ERK), protein kinase C (PKC), Akt (PKB), rapamycin ở người (mTOR), và glycogen synthase kinase 3 (GSK3). Sau khi tiêm truyền TNF-α ở người khỏe mạnh, kháng insulin phát triển trong cơ xương được liên kết với phosphoryl suy thoái của Akt nền 160, dẫn đến rối loạn chức năng vận chuyển glucose 4 (GLUT4) chuyển vị và sự hấp thu glucose. Nồng độ quá mức của TNF-α điều tiết tiêu cực đến tín hiệu insulin và sự hấp thu glucose toàn bộ cơ thể ở người. Ngoài ra, TNF-α kích thích phân hủy mỡ và giải phóng axit béo tự do. Axit béo tự do gây ra tích tụ nội bào diacylglycerol và acyl CoA chuỗi dài, tiếp theo protein kinase C kích hoạt và hủy phosphoryl hóa của IRS-1/2 trong tế bào cơ xương của người. Các nghiên cứu khác chứng minh rằng IL-6 cũng có thể ức chế lộ trình truyền tín hiệu insulin ở cấp độ các thụ thể insulin và tương tác IRS-1. Theo dõi một chất đối kháng thụ thể IL-1 ở những bệnh nhân với tiểu đường typ II không chỉ làm giảm CRP huyết thanh và IL-6 mà còn cải thiện độ nhạy insulin. Khiếm khuyết trong tín hiệu insulin thông qua các protein kinase Akt làm cơ sở cho sự phát triển của KI. Akt đóng vai trò như một điểm nút trong sự kiểm soát của cả trao đổi chất và tác dụng đa hiệu của insulin.
Fetuin-A, còn được gọi là a2-Heremans-Schmid glycoprotein (Ahsg), là một chất ức chế mạnh quá trình vôi hóa mạch máu. Bệnh nhân bệnh thận mạn có nồng độ fetuin-A thấp hơn đáng kể so với người khỏe mạnh. Điều trị ngắn hạn với sevelamer làm tăng fetuin-A huyết thanh tập trung trong bệnh nhân suy thận giai đoạn 4. Ở những bệnh nhân lọc máu, điều trị đồng vận canxi cho cường cận giáp thứ phát làm giảm đáng kể mức độ fetuin-A. Tuy nhiên, mức fetuin-A huyết thanh tăng lên rõ ràng sau khi cắt bỏ tuyến cận giáp ở bệnh nhân cường cận giáp. Fetuin-A ức chế hoạt động của thụ thể insulin tyrosine kinase bằng cách chặn sự tự động phosphoryl hóa của tyrosine kinase và IRS-1 và gây ra viêm cấp độ nhẹ, kết quả gây ra KI. Fetuin-A trong huyết thanh có liên quan tới KI và các bệnh đi kèm của nó, chẳng hạn như hội chứng chuyển hóa và đái tháo đường typ 2. Nồng độ fetuin-A cao hơn gắn liền với đái tháo đường typ 2 và KI ở bệnh nhân trung niên và cao tuổi. Tuy nhiên, những tác động có thể có của fetuin-A trên KI trong BTM cần được làm sáng tỏ hơn nữa.
Bộ ức chế các tín hiệu cytokine (SOCS) là một họ các protein trong tế bào, một số trong đó nổi lên với vai trò điều chỉnh sinh lý quan trọng của cân bằng nội môi thông qua cytokine, bao gồm miễn dịch bẩm sinh và thích ứng. Ở bệnh thận mạn hoặc bệnh thận giai đoạn cuối, sự gia tăng của SOCS trong bạch cầu đơn nhân và tế bào lympho, kèm theo tăng nồng độ trong huyết tương của các cytokine viêm IL-6, CRP, và TNFα, đã được ghi nhận. Quan sát gần đây cũng cho thấy mức độ biểu hiện của SOCS cũng thay đổi sâu sắc trong suy thận.
Protein SOCS hoạt động như một vòng phản hồi cổ điển để đảm bảo điều chỉnh lộ trình tín hiệu cytokine, và mục tiêu kinh điển của các protein SOCS là sự ức chế con đường tín hiệu JAK-STAT. Protein SOCS cũng có thể ức chế tín hiệu insulin; SOCS-1 ở chuột bị ngất cho thấy hạ đường huyết và tăng tín hiệu insulin. Các nghiên cứu gần đây cho thấy rằng các protein SOCS không chỉ làm trầm trọng thêm KI mà còn đóng vai trò như một cầu nối quan trọng giữa các cytokine cấp cao và KI. SOCS-3 có thể buộc Tyr960 phosphoryl hóa các thụ thể insulin, mà quan trọng là trên IRS-1 gắn kết. Ức chế SOCS-1 thường thông qua trung gian liên kết với các miền kinase của thụ thể insulin, ngăn ngừa sự phosphoryl hóa hơn nữa .
IL-6 tăng cao gây ra từ SOCS-3 và do đó ức chế tín hiệu insulin trong sợi cơ người phân hóa được nuôi trong ống nghiệm. SOCS-3 biểu hiện không cao trong cơ ở bệnh nhân béo phì không đái tháo đường và đái tháo đường typ 1. Tăng SOCS-3 biểu hiện ở những bệnh nhân đái tháo đường typ 2 có thể được giải thích bởi sự kết hợp của nồng độ glucose và IL-6 trong máu cao. Hàm lượng cao các cytokine gây viêm dẫn đến tăng SOCS-1 và SOCS-3 biểu hiện ở các mô nhạy cảm insulin, gây nên KI thông qua sự ức chế của con đường truyền tín hiệu insulin.
2.2. Vai trò của chuyển hóa mỡ
Chuyển hóa mỡ trong huyết tương cao rõ rệt ở bệnh nhân tăng urê máu, chủ yếu do giảm bài tiết qua thận. Béo phì cộng thêm tăng urê máu có thể làm trầm trọng thêm rối loạn mỡ máu. Mức tế bào mỡ bất thường, bao gồm leptin và adiponectin, có thể thúc đẩy hơn nữa KI và một trạng thái tiền viêm trong bệnh thận mạn. Leptin có thể được coi như là một chất độc urê máu và có liên quan tới giảm năng lượng nạp vào và suy dinh dưỡng protein năng lượng ở bệnh nhân tăng urê máu. Leptin cũng ngăn cản bạch cầu trung tính hóa hướng động và gây tổn thương mạch máu thông qua xơ vữa động mạch và hiệu ứng tiền viêm. Adiponectin có thể làm trung gian cho các hoạt động kích hoạt insulin, chống vữa xơ động mạch, chống viêm. Tín hiệu adiponectin qua MAPK quy định rối loạn oxy hóa, phân đoạn phản ứng tổng hợp ở tế bào có chân, và albumin niệu. Ở những bệnh nhân chạy thận nhân tạo, nồng độ adiponectin huyết thanh tỷ lệ nghịch với chỉ số khối lượng cơ thể, nồng độ insulin, và chỉ số HOMA.
Ở người, resistin có mặt ở các đại thực bào cư trú trong các mô mỡ. Resistin xuất hiện làm xáo trộn phản ứng miễn dịch và góp phần làm tăng nguy cơ xơ vữa động mạch bằng cách điều khiển các hoạt động của các tế bào nội mô. Mức độ lưu hành resistin có liên quan nhiều đến tốc độ thanh lọc glucose và viêm đánh dấu sinh học trong bệnh thận mạn, nhưng không phải với KI. Visfatin chủ yếu được tổng hợp trong mỡ nội tạng và có liên quan đến suy giảm chức năng nội mô và sự hình thành mạch. Ở những bệnh nhân LMB, visfatin huyết thanh có thể là một dấu hiệu nhạy cảm đối với hoạt động của tim. Chemerin là một adipokine mới tìm thấy, liên quan đến bệnh béo phì và cũng tăng ở bệnh nhân tăng ure. Ở những bệnh nhân chạy thận nhân tạo, chemerin cao có liên quan tới khả năng sống sót bất kể tương quan thuận của nó với các dấu hiệu của chứng viêm và rối loạn lipid máu.
Nghiên cứu trước đây chỉ ra rằng các cytokine tiền viêm có nguồn gốc từ mỡ phát huy tác dụng ức chế trực tiếp tín hiệu insulin cơ, gây ra sự liên quan chặt chẽ giữa KI và viêm hệ thống. Béo phì hoặc tích tụ mỡ ở vùng bụng có liên quan với sự rối loạn oxy hóa và suy giảm hoạt động của insulin ở BTM. Có một mối quan hệ chặt chẽ giữa các chỉ điểm sinh học của viêm và khối lượng chất béo ở bệnh nhân bệnh thận giai đoạn cuối. Giảm cân có thể giảm gánh nặng viêm và oxy hóa trong bệnh thận mạn và làm giảm bớt nguy cơ tim mạch ở các bệnh nhân này.
2.3. Các yếu tố khác
Có bằng chứng cho thấy trục vitamin D và hormone tuyến cận giáp (PTH) đóng vai trò quan trọng trong sinh lý bệnh của không dung nạp glucose và KI ở bệnh nhân suy thận. KI có mặt trong giai đoạn đầu của suy thận và có một liên quan nghịch với 25(OH)- vitaminD3. Chuyển hóa vitamin D bị rối loạn ở bệnh nhân suy thận mạn; bất thường bắt đầu trong suy thận các giai đoạn đầu, tức giai đoạn 3 hoặc sớm hơn, và tăng dần theo suy giảm chức năng thận. Vitamin D được công nhận là có vai trò đối với xương và cân bằng khoáng nội môi, thụ thể vitamin D và hệ thống chuyển hóa xác định trong nhiều mô. Nồng độ lưu thông 25-hydroxyvitamin D tương quan với sự dung nạp glucose, chức năng của tế bào beta, và độ nhạy insulin, được đo bằng các xét nghiệm dung nạp glucose và kẹp tăng đường huyết. Sau 4 tuần điều trị 1,25 (OH)2D3 tĩnh mạch, tình trạng không dung nạp glucose, KI, giảm insulin, và tăng triglyceride máu đã được cải thiện ở bệnh nhân lọc máu chu kỳ, trong sự vắng mặt của PTH ức chế.
Sự tích tụ các chất độc urê máu có thể góp phần gây ra KI trong bệnh thận giai đoạn cuối. Các peptide có trọng lượng phân tử trung bình trong huyết thanh tăng ure gây ra KI trong các tế bào mỡ. Pseudouridine được tích lũy trong hệ tuần hoàn, và dimethyl arginine bất đối xứng (ADMA) được công nhận là một chất độc urê máu quan trọng liên quan đến bệnh thận mạn và KI.
Toan chuyển hóa cũng có thể đóng một vai trò quan trọng với KI trong suy thận mạn. Nó có liên quan đến suy dinh dưỡng protein năng lượng và viêm. Bằng chứng cho thấy rằng những tác động dị hóa của toan chuyển hóa có thể do tăng hoạt động của ubiquitin-proteasome phụ thuộc adenosine triphosphate (ATP) và chuỗi nhánh ketoacid dehydrogenase. Điều trị nhiễm toan chuyển hóa với bicarbonate uống làm tăng cả độ nhạy cảm insulin và bài tiết insulin ở bệnh nhân suy thận mạn, được xác định bằng nghiệm pháp kẹp đẳng đường cường insulin
Bệnh nhân bệnh thận giai đoạn cuối thường có sức đề kháng của cơ thể thấp, điều này có thể góp phần vào KI và rối loạn chuyển hóa. Cải thiện đáng kể dung nạp glucose và độ nhạy insulin đã được tìm thấy ở những bệnh nhân bệnh thận mạn sau 6 hay 9 tháng điều trị EPO cho bệnh thiếu máu. Cải thiện khả năng đề kháng sau khi điều trị EPO có thể là yếu tố quan trọng trong việc cải thiện quá trình chuyển hóa glucose ở bệnh nhân suy thận.
Kết luận
KI phổ biến ở các bệnh nhân BTM và đóng một vai trò trong suy giảm chức năng thận. Các nguyên nhân của KI là đa yếu tố và có liên quan đến một mạng lưới phức tạp bao gồm cả viêm mãn tính, rối loạn oxy hóa, thiếu vitamin D, thiếu máu, suy dinh dưỡng. Những yếu tố này được kết hợp với các cytokine viêm, chuyển hóa mỡ, rối loạn ER, và SOCS, dẫn đến một khiếm khuyết mắc phải của con đường tín hiệu thụ thể insulin.
(Lược dịch từ Insulin Resistance in Patients with Chronic Kidney Disease, Journal of Biomedicine and Biotechnology Volume 2012, Article ID 691369, của tác giả Min-Tser Liao, Chih-Chien Sung, Kuo-Chin Hung, Chia-Chao Wu, Lan Lo, Kuo-Cheng Lu ).
- 28/03/2016 21:49 - Chảy máu và các rối loạn đông máu trong ICU (p.1)
- 28/03/2016 21:32 - Dò động mạch cảnh – xoang hang
- 17/03/2016 19:34 - Các dấu hiệu CT trong thủng ruột non
- 06/03/2016 15:45 - Sử dụng Intralipid 20% trong điều trị ngộ độc thuố…
- 25/01/2016 14:41 - Điều trị kháng sinh theo chỉ dẫn procalcitonin cho…
- 25/01/2016 10:11 - An toàn tim mạch của các thuốc kháng viêm nonstero…
- 16/01/2016 19:12 - Lọc máu trong nhiễm trùng huyết
- 16/01/2016 18:33 - Những điều răn từ Guidelines ESC 2015
- 16/01/2016 16:47 - Điều trị kháng sinh theo chỉ dẫn procalcitonin cho…
- 04/01/2016 09:21 - Sử dụng test VIA trong tầm soát ung thư cổ tử cung





